1:デュシェンヌ(Duchenne)型筋ジストロフィー (DMD:Duchenne muscular dystrophy)
(1)概要
①原因
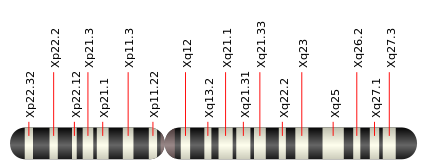
X染色体短腕Xp21.2にある、ジストロフィン遺伝子の異常によるジストロフィノパチーです。
小児筋ジストロフィーでは最重症となります。
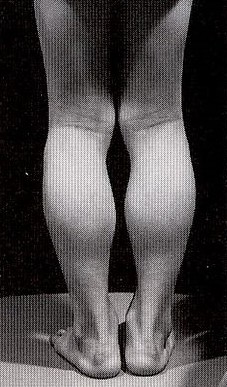
②特徴
腓腹筋肥大が特徴です。
仮性肥大(筋線維が脂肪や結合組織に変性)の場合も有ります。
ほとんどは知的障害がありません。
寿命---30歳前後です。
死因---呼吸不全70~80%、心不全10~20%。
(2)原因
①病因
ジストロフィンの完全欠損により発症します。
ジストロフィンは棒状の細胞質タンパク質で、コスタメアとして知られるタンパク質複合体の一部をなしています。
この複合体は細胞膜を越えて、筋繊維の細胞骨格とその周囲の細胞外マトリックスを接続しています。
通常の骨格筋組織は少量の(全タンパク質の0.002%)ジストロフィンを含んでいます。
その欠損・変異は、細胞内シグナル伝達系の異常をもたらし、不可逆的な筋繊維壊死・筋力低下・疲労などの症状を
引き起こす原因となります。
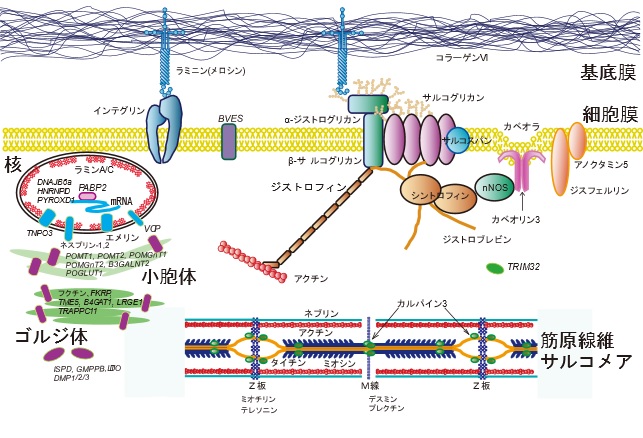
「難病情報センター HP」 から引用
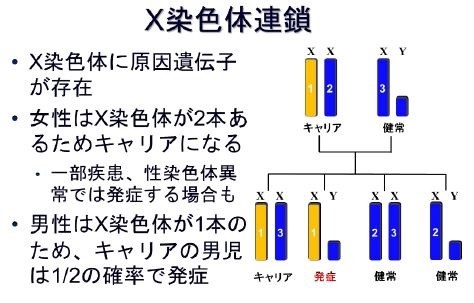
②遺伝様式
X連鎖劣性遺伝形式で、基本的には男児に発症します。
まれに染色体異常により女性にも発症する事があります。
2/3は保因者である母親からの遺伝です。
1/3は家族歴を欠く新規突然変異により発症します。
補足:遺伝形式---X連鎖劣性遺伝
X染色体上にある遺伝子の変化によって発症する疾患です。
女性はX染色体を2本、男性はX染色体を1本持っています。
通常男性のみ発症します。
女性は、変化のない正常なX染色体も持っているため、発症しても男性の患者さんよりは軽症です。
男性患者さんのお母様は、遺伝子変異をもつ保因者である可能性があります。
3:症状
(1)経年的な的症状の変化
生下時には異常は有りませんが、経年的に症状が出てきます。
①2歳
この頃までは症状が無く、歩行可能です。
②2~4歳頃
転びやすい、走れない、階段が昇りにくいなどが確認されるようになります。
初期には腰帯筋、次第に大殿筋、肩甲帯筋へと筋力の低下の範囲を広げていきます。
なお、筋力低下は対称的に起きるという特徴を持ちます。
③3~10歳
1)動揺性歩行(waddling gait)
動揺性歩行(アヒル歩行)となります。
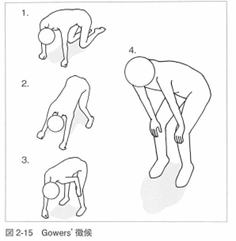
2)ガワーズ(Gowers)徴候
膝に手をついて自分の体によじ登るような 立ち上がり方をします。
登攀性起立ともいいます。.
仮性肥大、腓腹筋肥大があります。
④10歳前後
歩行困難となり、車椅子生活になります。
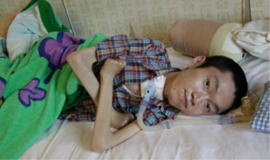
⑤18歳前後
寝たきりとなります。
呼吸筋の障害により、気管切開・人工呼吸器装着も必要となります。
⑥30歳前後
心筋疾患を合併することが多く、心不全は大きな死因のひとつです。
30歳前後で死亡します。
4:診断
(1)診断
男児で腓腹筋肥大→ジストロフィノパチーを疑います。
確定診断は遺伝子検査(MLPA法) で確定します。
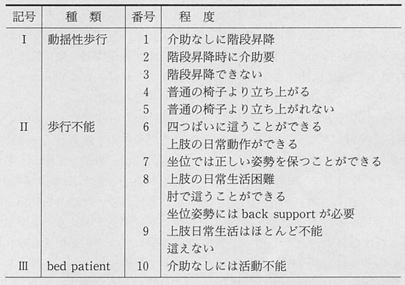
(2)進行性筋ジストロフィー症 機能障害度分類(厚労省)
5:治療
(1)根治的治療
根本的治療は有りません。
(2)対症療法
機能訓練や関節拘縮予防のためのストレッチ(理学療法)のほか、心不全・呼吸障害に対する対症療法が行われます。
少量の副腎皮質ステロイド内服で歩行可能期間延長のエビデンスが有り、保険適用されています。
2:ベッカー(Becker)型筋ジストロフィー (BMD:Becker muscular dystrophy)
(1)概念
①原因
DMDと同じジストロフィノパチーです。
② 特徴
病態はデュシェンヌ型と同じですが発症時期が遅く、症状の進行も緩徐、関節拘縮も少ないタイプです。
一般に予後は良好です。
発症年齢が遅く歩行可能期間も長い良性の経過をたどる病型です。
発症年齢は小児から高齢者まで様々です。
(2)症状
①主要症状
腓腹筋肥大
病初期からしばしばふくらはぎの痛みが有ります。
筋症状は軽度ですが、高度の心筋症合併することもあります。
②DMDとの区別
15歳で歩行可能であるかが目安です。
DMD---15歳で歩行不可。
BMD---15歳で歩行可能。
デュシェンヌ型ではジストロフィン蛋白がほとんど発現していません。
これに対し、ベッカー型では異常なジストロフィン蛋白が産生されたり、発現量が少ないことが知られています。、
これにより両者の症状の差異が生じているのだと考えられています。
(3)診断
①検査
検査としては、血液検査、筋病理検査、遺伝子検査などがあります。
筋肉の破壊を血液検査で確認することも可能です。
遺伝子検査(MLPA法)
(4)治療
①根治療法
根本的治療法は有りません。
②対症療法
歩行が難しくなった場合には車いすを使用します。
心不全を発症した場合には、必要に応じた治療薬を選択します。
3:エメリー・ドライフス型筋ジストロフィー (EDMD:Emely-Dreifuss muscular dystrophy)
(1)概念
①特徴
幼小児期に発症する筋ジストロフィー、比較的緩徐に進行します。
筋力低下のほか、比較的早期から認める関節拘縮、心伝導障害を主症状とする疾患です。
(2)原因
①病因
核膜蛋白であるエメリン、もしくはA型ラミンの遺伝子変異による遺伝性筋疾患です。
②遺伝様式
X連鎖劣性遺伝のEDMD1と、常染色体優性遺伝のEDMD2があります。
1)EDMD1
Xq28にあるエメリン遺伝子(EMD)変異により、核膜蛋白エメリンの欠損により発症します。
2)EDMD2
1番染色体1q21.2上の核膜マトリックス蛋白ラミンA/Cの遺伝子(LMNA)変異により発症します。
(3)疫学
発生頻度は10万人あたり0.1未満です。
好発年齢は2~10歳の乳幼児から成人です。
(4)症状
次の3つが特徴的な症状です。
①上腕―腓骨型の筋委縮・筋力低下
②肘・足関節・後頚部の早期関節拘縮
関節拘縮は筋力低下前からみられます。
傍脊柱筋が侵され脊椎強直症候群(rigid spine syndrome)を呈します。
③心伝導障害
進行は緩徐ですが、致死的不整脈による突然死のリスクがあります。
(5)治療・予防
①根治的療法
根本的治療はない.
②予防的治療
心伝導障害に対して早期の心臓ペースメーカー挿入で突然死予防が可能斗なります。
4:顔面・肩甲・上腕型筋ジストロフィー (FSHD:Facio-scapula-humeral dystrophy)
(1)概念
①特徴
顔面、肩甲帯、上腕の筋力低下・筋萎縮が特徴です。
進行すると全身の筋力低下が見られるようになります。
(2)原因
①DUX4遺伝子の発現
酸化ストレスが顔面肩甲上腕型筋ジストロフィーの原因遺伝子DUX4を増加させます。
4番染色体長腕の端(4q35)にある、成人では通常発現していないDUX4遺伝子が発現するようになり、
筋細胞を障害することが本症の本質的な原因と考えられています。
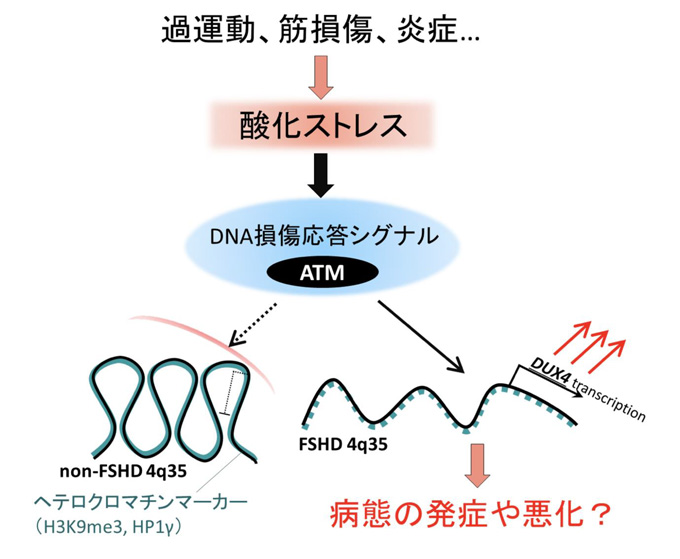 「京都大学iPS細胞研究所HP」から引用 「京都大学iPS細胞研究所HP」から引用
「A Patient-derived iPSC Model Revealed Oxidative Stress Increases Facioscapulohumeral
Muscular Dystrophy-causative DUX4」
(3)疫学
①有病率
10万人に2人位です。
遺伝性筋疾患中でDMD、筋強直性筋ジストロフィーに次いで多い病気です。
10~20歳に発症します。
②遺伝様式
常染色体優性遺伝.
約30%は新規突然変異による孤発例.
(4)症状
①特徴
しばしば筋萎縮や筋力に著しい左右差が見られます。
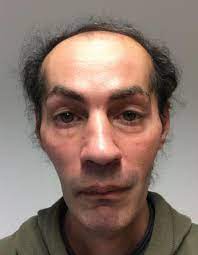
顔面筋が侵されミオパチー顔貌を呈します。
ただし咬筋や咽頭筋、舌筋は侵されないので構音や嚥下機能は正常です。
呼吸障害や心筋障害の合併はなく、予後良好です。
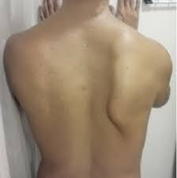
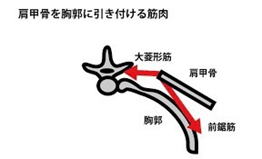
②翼状肩甲
肩甲骨付近の筋肉萎縮により翼状肩甲を呈します。
③Beevor徴候
腹筋を使って起き上がるとき、臍が上方に移動するBeevor徴候を呈します。
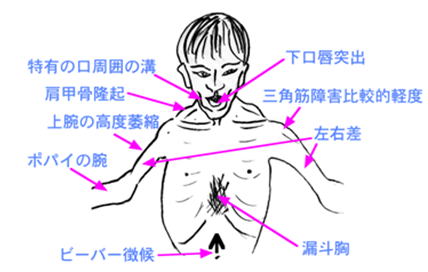
(5)診断
特徴的な罹患筋分布と、症状の左右差が診断の手がかりとなります。
5:肢帯型筋ジストロフィー( LGMD :Limb-girdle muscular dystrophy)
(1)概念
①特徴
1歳以後に体幹に近い部分の筋肉(近位筋)の筋力低下で生じる筋ジストロフィーの総称です。
特有な罹患筋分布や特徴的な臨床像を欠き、腰帯や肩甲帯の筋萎縮で発症するものです。
(2)分類
①LGMD1型
常染色体顕性(優性)遺伝形式を示します。
一組の遺伝子の一方に変異があれば発症するものです。
②LGMD2型
常染色体潜性(劣性)遺伝形式
一組の遺伝子の両方に変異がある場合にのみ発症するものです。
(3)原因
LGMDのうち約60パーセントは、まだ原因となる遺伝子やタンパク質が同定されていません。
様々な遺伝子転座や原因遺伝子が特定されてきています。
(4)疫学
日本での患者数は、2000人程度dす。
LGMD1= 5%
LGMD2=95%
(5)症状
青年期に腰帯筋の筋萎縮・筋力低下で発症.
進行とともに上肢の筋萎縮.
DMD様の重症例から軽症例まで幅広い.
呼吸障害と心筋障害の有無が生命予後の重要因子.
(6)診断
①確定診断
各々の病型を臨床徴候のみでの診断は不可能、筋生検が必要です。
確定診断は遺伝子検査です。
②鑑別診断
代謝性ミオパチー、炎症性ミオパチー、脊髄性筋萎縮症などが鑑別対象となります。
(7)治療
①治療・療養上の注意
定期的な経過観察を行い、合併症に注意する事が重要です。
病状の進行と合併症に注意した適切な医療によって、生存期間が延長し、生活の質が向上します。
筋力低下に対しては、症状に合わせてリハビリテーションを受け、拘縮が起きないようにします。
②注意するべき合併症
病型によって異なりまが、とくに下記の合併症が考えられる病型では確実な経過観察が必要です。
1)心不全(心筋障害)
注意深く経過観察を行い、必要に応じてACE阻害剤やβ-遮断薬投与、ペースメーカーの使用考えます。
2)呼吸不全(呼吸障害)
定期的に呼吸機能評価を行い、必要に応じて呼吸理学療法、人工呼吸器の使用を考えます。
6:先天性筋ジストロフィー (CMD:Congenital muscular dystrophy)
(はじめに)
生下時ないし乳児期早期から筋緊張低下・筋力低下(floppy infant)を呈する病型のMDです。
脳奇形を合併するタイプと、筋ジストロフィー病変のみのタイプに大別されます。
前者の代表が我が国特有の福山型先天性筋ジストロフィーです。
福山型(FCMD)
メロシン欠損型
ウルリッヒ(Ullrich)型
6-1:福山型先天性筋ジストロフィー (FCMD:Fukuyama Congenital muscular dystrophy)
(1)概念
日本人特有の常染色体劣性遺伝の疾患です。
筋ジストロフィーに加え、脳奇形と眼病変の合併が特徴です。
(2)原因
9番染色体長腕9p31にあるフクチン遺伝子の変異で発症します。
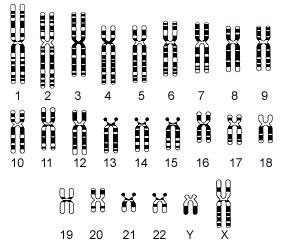
(3)疫学
我が国の小児筋ジストロフィー中、DMDに次いで二番目に多い病気です。
有病率=3人/10万人 位です。
(5)症状
①特徴的な症状
典型例は頚定の遅れや、運動発達遅滞をおこします。
重症例では生下時から呼吸不全、哺乳力低下芽あります。
筋トーヌスの低下を示す乳児の総称であるfloppy infantともいわれます。
顔面筋罹患のため、表情に乏しく開口していて高口蓋です。
早期から関節拘縮をお越し、進行で全身の関節拘縮へと進みます。
知的障害を必発します。(IQ50を超えることはまれ)
けいれん(約50%)が併発します。
喀痰の排出困難により肺炎を起こしやすくなります。
②顔貌・口腔の特徴
常に開口・開咬を呈します。
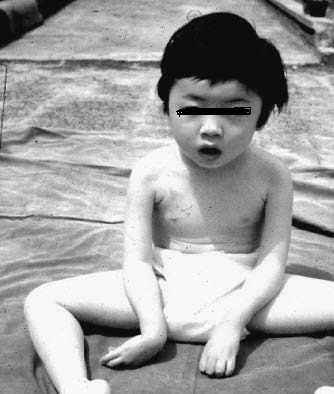
(6)診断
①確定診断
フクチン遺伝子検査により確定診断が得られます。
②鑑別診断
筋症状、脳奇形、眼病変が共通する疾患にWalker-Warburg症候群との鑑別が必要です。
(7)経過・予後
呼吸筋力低下による呼吸不全,拡張型心筋症による心不全,嚥下障害による誤嚥や窒息が予後を左右します。
平均寿命は10歳代後半から20歳代前半です。(15歳程度)
(8)治療
有効な治療はなく、リハビリテーションなどの対症療法を行います。
6-2:メロシン欠損型筋ジストロフィー
(1)概念
欧米に頻度が高く日本では稀な筋ジストロフィー症です。
ラミニンα2鎖をコードするLNMA2遺伝子により、ラミニン211(メロシン)の完全または部分欠損により生じます。
非福山型に分類され,知的障害を伴わない先天性筋ジストロフィーです。
(2)原因
ラミニンは基底膜の主要な構成成分で,組織を区分し細胞浸潤を防ぐ役割を担っています。
α,β,γ鎖から構成される糖蛋白の三量体で,骨格筋に発現するのはα2,β1,γ1鎖からなるラミニン211であり、
別名メロシンと呼ばれています。
メロシンはα-DGの糖鎖やインテグリンと結合することで基底膜と連結固定しています
染色体6q22-q23に存在するラミニンα2鎖をコードするLNMA2遺伝子変異により,メロシンの完全または部分欠損
により生じます。
(3)疫学
欧米では非福山型(古典型)の約半数はメロシン欠損型とされますが、本邦での報告は非常に稀です。
(4)症状
①全欠損型
臨床症状は重症で、出生時よりの哺乳困難、啼泣微弱、著明な筋緊張低下、筋力低下、顔面筋罹患、多関節拘縮、
を認めます。
出生時より呼吸障害はあるが,換気補助を必要とするほどの呼吸障害は,遅れて乳幼児期に発症します。
最高運動到達は通常独坐か支え立位で,歩行可能例は少ない.
進行性に関節変形,拘縮,側弯が生じる.腱反射は早期に消失します。
②部分欠損型
発症が遅く,肢帯型筋ジストロフィーに類似した緩徐進行性の筋力低下を示します。
心合併症は非常に稀です。
約30%の例でてんかんの発症を認め,知能は多くは正常から境界域です。
(5)治療
対症療法が中心です。
管理上一番の問題は進行性呼吸障害です。
10歳未満で30%は夜間の呼吸補助が必要となることから,早期からの呼吸モニタリングが必要です。
呼吸理学療法,非侵襲的陽圧換気療法,器械的咳介助を必要に応じて導入します。
理学療法,進行する関節拘縮,側彎への整形外科的フォローも重要です。
時に哺乳障害,体重増加不良に対して,経管栄養,胃瘻造設が必要となります。
(6)予後
緩徐進行性であり,心筋症の合併が少ないため比較的予後は良好です。
一方で、呼吸不全により新生児期,乳児期早期に死亡する例もあります。
6-3:ウルリッヒ(Ullrich)型筋ジストロフィー症
(1)概念
日本では福山型先天性筋ジストロフィーの次に頻度の高い先天性筋ジストロフィーです。
VI型コラーゲンの異常より,重症型のUllrich型先天性筋ジストロフィーと軽症型のBethlem型ミオパチーを生じます。
(2)原因
Ullrich型はVI型コラーゲンをコードする3つの遺伝子の変異により発症します。
主に常染色体劣性遺伝をとるが優性遺伝もあり得ます。
筋組織の免疫組織学的検討ではVI型コラーゲンの完全欠損を示す場合と筋鞘膜特異的欠損を示す場合があります。
(3)疫学
福山型先天性筋ジストロフィーの次に頻度の高い先天性筋ジストロフィーとされています。
希少疾病ではあるものの一定数の患者が存在するものと考えられます。
(4)症状
新生児期よりの筋緊張低下,哺乳障害に加え,頸・肩・肘・股・膝関節などの近位関節の拘縮と手・足・指関節の
過伸展という特徴的な所見を示します。
斜頸,踵骨の突出,先天性股関節脱臼も多く,独歩可能な例は半数にとどまります。
典型例では10歳までに歩行不能となるが,20歳を過ぎても歩行可能な軽症例も存在します。
皮膚は過伸展し,発汗過多に加え,粗な小胞性角化症,紙やすり様丘疹を認めます。
創傷治癒が遅く,ケロイドが残ることも特徴の一つです。
顔面筋罹患を認め,高口蓋,突出した耳介を伴い,丸く特徴的な顔貌を示します。
知能は通常正常です。
脊柱の可動域制限と側彎は早期から出現します。
(5)治療
現時点では対症療法に限られます。
特に慢性呼吸不全は必発であり,呼吸管理が重要になります
小児期から定期的に呼吸モニタリングを行い,呼吸理学療法と非侵襲的陽圧換気療法を必要に応じて導入します。
(6)予後
呼吸筋力低下による呼吸不全が予後を左右します。
7:ミオトニア症候群
(はじめに)
筋強直(myotonia)とは、収縮した筋肉が弛緩しにくい現象の事です。
筋強直現象を呈する疾患は筋強直症と総称されます。
次の二つにに大別されます。
筋強直性ジストロフィー
非ジストロフィー性筋強直症
7-1:筋強直性ジストロフィー(DM:dystrophia mytonica)
(1)概念
常染色体劣性遺伝疾患.
筋力低下・筋萎縮に加え筋強直症が特徴だが、多臓器障害により多彩な症状.
母親由来が多い.
(2)原因
DM1とDM2の二つがありますが、ほとんどはDM1です。
①DM1
DM1は19番染色体長腕19q13.3にあるDMキナーゼ遺伝子のCTG繰り返し配列の異常伸長により発症します。
②DM2
DM2はまれで、3番染色体3q21上のCCTG繰り返し配列の異常伸長により発症します。
(3)疫学
患者数は10万人あたり7人程度で、日本に10,000人程度の計算にると推計されています。
原因となる遺伝子によって1型と2型に分類されますが、日本ではほとんどが1型です。
DM2は本邦では1家系のみ同定されています。
成人発症の筋ジストロフィーでは最多です。
(4)症状
主要症状
筋強直(ミオトニア現象)と、筋萎縮、筋力低下が主徴です。
通常、筋疾患では近位筋が侵されやすいのですが、DM1は遠位筋優位となります。
筋強直(ミオトニア現象:収縮筋が弛緩しにくい)
進行性の筋萎縮と筋力低下
多臓器障害
①筋強直(ミオトニア現象)
収縮筋が弛緩しにくい現象です。
握った手指が開きにくい (把握性筋強直:grip myotonia)などが特徴的です。
  
②進行性の筋萎縮と筋力低下
咬筋・胸鎖乳突筋の筋萎縮、側頭筋の筋萎縮(白鳥の頸)、肢遠位筋の筋萎縮が起こります。
その結果、フタが開けにくい、仰向けからすぐに起き上がれない、口笛が吹けない、等の状態になります。

③多臓器障害症状
そのほかにも白内障や知能低下、心伝導障害などの多彩な症状を併発します。
呼吸器症状(呼吸筋麻痺、排痰能力低下、睡眠時無呼吸)
心機能異常(不整脈、心筋障害)
中枢神経異常(認知機能低下、性格変化、日中過眠症)
消化器症状(消化管拡張、便秘、下痢、腸閉塞、肝機能異常、胆石症)
眼症状(白内障、網膜色素変性症、眼瞼下垂)
耳鼻科症状(難聴、中耳炎、副鼻腔炎、嚥下障害)
内分泌異常(耐糖能異常、脂質異常症)
生殖機能低下(性腺機能異常、不妊症)
腫瘍ができやすい、など。
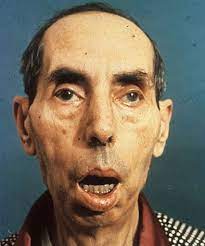
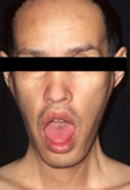
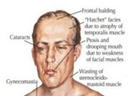
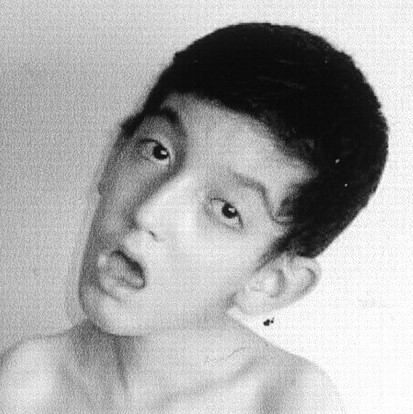
④特徴的顔貌
咬筋、側頭筋、顔面筋の萎縮、こけた頬。
オノ様顔貌、眼瞼下垂、仮面様顔貌、開咬。前頭部禿頭(トクトウ)
(5)診断
①臨床診断
特有な顔貌(斧様顔貌)、罹患筋パターン、ミオトニア現象で臨床診断がなされます。
②確定診断
ミオトニアが明らかでなければ、筋電図検査、または遺伝子検査で確定診断とします。
(6)経過・予後
症状は10歳以降に現れ、きわめて緩徐に進行するため、発症時期の特定困難.
発症後30年以内で約半数が歩行不能.
生命予後は心筋症などの有無により変わる.
(7)治療
根本的治療はなく、リハビリテーション.
ミオトニアが著しく生活への支障をきたす場合、フェニトイン、カルバマゼピン、メキシレチンなどの薬物治療
が行われます。
|




