| その他の症候群 |
その他の症候群
Ⅰ:レット(Rett)症候群 (Rett syndrome)
Ⅱ:22q11 .2欠失症候群 (第三・第四鰓弓症候群)
Ⅲ:プラダー・ウィリ(Prader-Willi 症候群) (Prader-Willisyndrome)
Ⅳ:低リン血症性(ビタミンD抵抗性)くる病 (hypophosphatemic)
Ⅴ:もやもや病 (Moyamoya disease)
Ⅵ:レノックス・ガスト(Lennox-Gastaut)症候群 (Lennox-Gastaut syndrom)
Ⅶ:ウエスト(West)症候群 (West syndrome)
Ⅷ:ゴールデンハー症候群
Ⅸ:ラッセル・シルバー症候群(RSS)
Ⅹ:ポイッツ・イェガース(Peutz-Jeghers)症候群
|
| Ⅰ:レット(Rett)症候群 (Rett syndrome) |
1:概念
(1)特徴
重度知的能力障害、後天性小頭症、
手の機能的動作消失と特異な常同運動、
特徴的歩行障害が特徴的な症候群です。
 
(2)歴史
1966年にA.Rett (1924~1997)が報告しました。
(3)原因遺伝子と遺伝形式
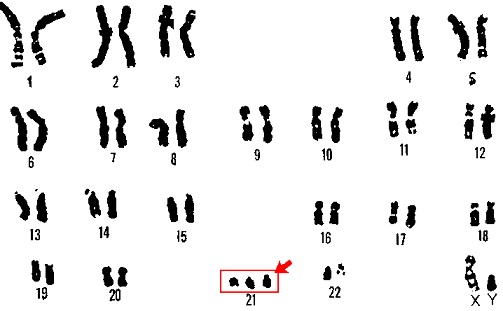
①典型例
80~90%にはX染色体(Xq28)にあるメチルCpG結合タンパクMEcm遺伝子の変異がみられます。
②非典型例
Xp22にあるサイクリン依存性キナーゼ様CDKZ5遺伝子の異常や14番染色体上のFDXGIの異常が見られます。
2:疫学
(1)出生頻度
2万人に1人。 典型例は女性だけにみられます。
3:レット症候群の症候・症状
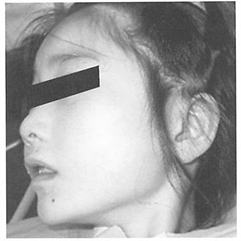
(1)全身的特徴
乳児前中期の筋緊張低や、乏しい表情などがあります。
乳児後期から自閉スペクトラム症に似た発達遅滞が出現します。
幼児期に小頭症と重度知的能力職害を示します。
1~2歳に特徴的常同運動(手もみ連動)が出現します。
体を左右に揺らした踏み出しと狭い歩幅での歩行があります。
 『スペシャルニーズデンティストリー障害者歯科 第2版』から引用 『スペシャルニーズデンティストリー障害者歯科 第2版』から引用
(2)合併症
心疾患---QT延長T波異常、心拍異常。
睡眠障害、側湾、間欠的過呼吸、無呼吸発作。
てんかん,発育不良、四肢末端の冷感、などがあります。
4:レット症候群と歯科医療
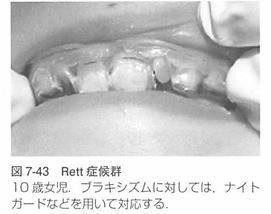
(1)歯科的特徴
ブラキシズム、歯列不正流涎、嚥下障害があります。
 『スペシャルニーズデンティストリー障害者歯科 第2版』から引用 『スペシャルニーズデンティストリー障害者歯科 第2版』から引用
(2)歯科的対応
ブラキシズムにはナイトガードなどを製作します。
てんかんに対応する必要が有ります。
無呼吸発作では、チアノーゼとともにSpO2が短時間で低下します。
必要に応じてモニタリングと酸素吸入を行います。
QT延長をきたす薬物を用いないようにします。
補足:薬剤性QT延長症候群を来たす薬剤
抗不整脈薬
キニジン、ジソピラミド(リスモダンR)、プロカインアミド(アミサリンR )、アミオダロン(アンカロンR )
ニフェカラント(シンビットR )、ソタロール(ソタコールR )、ベプリジル(ベプリコールR )など
向精神薬
アミトリプチリン(トリプタノールR ・・・三環系抗うつ薬) 、ハロペリドール(セレネースR )
クロルプロマジン(ウインタミンR ・コントミンR )、ピモジド(オーラップR )、チオリダジン など
抗菌薬
エリスロマイシン、クラリスロマイシン、スパルフロキサシン(スパラR )など
抗アレルギー薬
テルフェナジン(トリルダンR )、アステミゾール
その他
シサプリド 三酸化ヒ素
|
| Ⅱ:22q11 .2欠失症候群 (第三・第四鰓弓症候群) |
1:概念
(1)特徴
第三・第四鰓弓症候群とも呼びます。
胸腺低形成、先天性心疾患、特徴的顔貌、口蓋裂、低カルシウム血症によるテタニー、易感染性が特徴です。
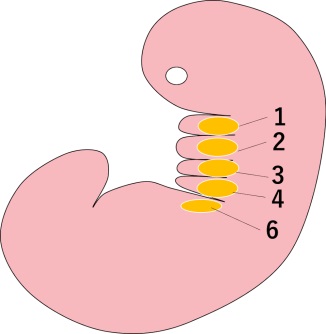
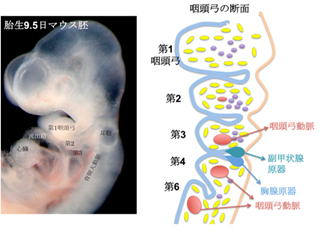
(補足)鰓弓
動物の初期発生で、魚類では鰓(エラ)に分化する部分に相当する領域です。
胎児期の初期に一時的に現れ、耳・鼻・咽頭・顎などの器官になります。
ヒトでは、咽頭や中耳などに分化し咽頭弓といいます。
ヒトの場合、咽頭弓は全部で6つあるが、第5咽頭弓は往々にしてほぼ欠如しているか、痕跡的です。
第1咽頭弓---顎骨弓
第2咽頭弓---舌骨弓
第3咽頭弓---舌咽神経(第Ⅸ神経)など
第4咽頭弓---迷走神経(第Ⅹ神経)の上咽頭枝、副神経、甲状軟骨など。
第6咽頭弓---
(2)歴史
1927年にWWernstedtが初めて報告し、1965年に米国の小児科医A.MDiGeorge (1921~2009)が詳細を報告し、
DiGeorge症候群としました。
現在では、円錐動脈幹異常顔貌症候群、口蓋心臓顔症候群など他の22q11.2の微細欠失とまとめられ、
22q11.2欠失症候群と呼ばれています。
(3)原因遺伝子と遺伝形式
22番染色体(22q11.2)のヘミ接合型(相I可染色体の片方のみ)欠失による症候群です。
ほとんどが散発性です。
特に、耳上皮や内耳周囲組織で発現するTBXI遺伝子のハプロ不全が原因とされています。
2:疫学
(1)出生頻度
4千人に1人.
3:22q11 .2欠失症候群の症候・症状
(1)全身的特徴
①特徴的顔貌
眼間開離、眼瞼裂斜下、耳介低位、短い人中、など。
②先天性心疾患
右側大動脈弓、総動脈幹遺残、 Fallot四徴症、大動脈狭窄症心室中隔欠損、大血管転位。
③胸腺不全に基づく細胞性免疫不全
④その他
軽度知的能力障害、鼻声、尿路奇形、など。
(2)合併症
水腎症、感染症、口腔内カンジダ症、下痢、慢性鼻炎,肺炎,髄膜炎、敗血症、など。
4:2q11 .2欠失症候群と歯科医療
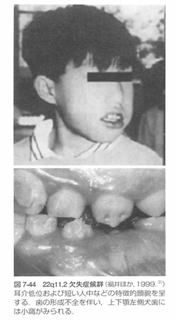
(1)歯科的特徴
小下顎症、二分口蓋垂、高口蓋、
エナメル質形成不全、歯の萌出遅延、
開咬や軟口蓋裂などがあります。
 『スペシャルニーズデンティストリー障害者歯科 第2版』から引用 『スペシャルニーズデンティストリー障害者歯科 第2版』から引用
(2)歯科的対応
エナメル質形成不全には、餓蝕予防処置を行います。
先天性心疾患への対応も行います。
|
| Ⅲ:プラダー・ウィリ(Prader-Willi 症候群) (Prader-Willisyndrome) |
1:概念
(1)特徴
新生児期の筋緊張低下と食欲抑制困難に伴う肥満などを特徴とする.
(2)歴史
1956年にA.Prader (1919~2001)とH.Willi(1900~1971) らが報告しました。
(3)原因遺伝子と遺伝形式
15番染色体の父親由来部位の15q11.2にあるSNPRN遺伝子、
およびニューロンの増殖や死を抑制するnecdine NDN遺伝子などの変異による隣接遺伝子症候群です。
2:疫学
(1)出生頻度
8千~2万5千人に1人.
3:プラダー・ウィリ症候群の症候・症状
(1)全身的特徴
新生児期の筋緊張低下、乳児期後半からの大食と肥満、
外性器低形成
知的能力障害および特徴的行動(澗艤や頑固さなど)など。
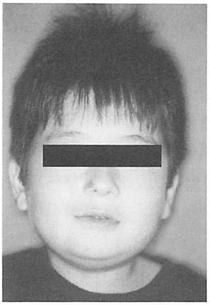
(2)特徴的顔貌
幅の狭い前額部、アーモンド型眼瞼裂、斜視、狭い鼻稜、U字型の唇紅・上口唇。
 『スペシャルニーズデンティストリー障害者歯科 第2版』から引用 『スペシャルニーズデンティストリー障害者歯科 第2版』から引用
(3)合併症
肥満に伴う糖尿病、呼吸器感染症、心疾患
Pickwickian症候群(強度の肥満を伴う一群) 、色素低下などがあります。
4:プラダー・ウィリ 症候群と歯科医療
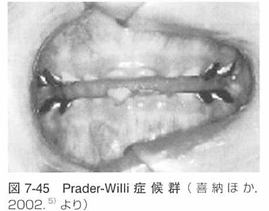
(1)歯科的特徴
エナメル質形成不全、不正咬合、歯の萌出遅延、
高口蓋,歯の数と形の異常.咬耗、
口腔乾燥や唾液の粘性があります。
 『スペシャルニーズデンティストリー障害者歯科 第2版』から引用 『スペシャルニーズデンティストリー障害者歯科 第2版』から引用
(2)歯科的対応
齪蝕予防処置を行う.
矯正治療も検討する知的能力障害にも対応する.
|
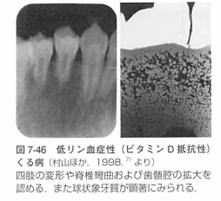
| Ⅳ:低リン血症性(ビタミンD抵抗性)くる病 (hypophosphatemic) |
1:概念
(1)特徴
X連鎖性低リン血症性くる病とも呼ばれています。
低リン酸血症、腎臓におけるリン酸塩再吸収、ビタミンD代謝不全のため天然型ビタミンDで完治しない成長遅滞.
くる病、骨軟化性骨疾患が特徴です。
(2)歴史
1937年にF.Albright (1900~1969)が報告しました。
(3)原因遺伝子と遺伝形式
X染色体(Xp22.11)にあるリン酸塩調節エンドペプチターゼ皮HEX遺伝子の変異。
リンの転送障害,
特に腎近位尿細管におけるリンの再吸収低下と腸管におけるリンの再吸収低下を生じます。
X連鎖性優性遺伝であり、家族内発症があります。
2:疫学
(1)出生頻度
2万人に1人.
3:低リン血症性くる病の症候・症状
(1)全身的特徴
四肢の変形、歩行異常、O脚あるいはX脚.
脊椎轡曲性骨変形~低身長。
くる病念珠(肋骨軟骨接合部拡大)、など。
(2)合併症
強直性脊椎炎、脊髄の狭窄、腱や靭帯の石灰化や関節炎などがあります。
4:低リン血症性くる病と歯科医療
(1)歯科的特徴
歯の萌出遅延、歯髄腔の拡大、歯の形成異常。
特に象牙質の形成異常として、幅広い象牙前質、球状象牙質、歯髄からエナメル象牙境に達する管状欠陥があります。
齪蝕や外傷を伴わなくても突発的な歯性の歯肉膿瘍を形成することがあります。
 『スペシャルニーズデンティストリー障害者歯科 第2版』から引用 『スペシャルニーズデンティストリー障害者歯科 第2版』から引用
(2)歯科的対応
軽度な齪蝕や咬耗の進行から歯髄炎になることがあります。
餓蝕予防および早期治療に努めます。
歯の萌出遅延についてはX線撮影により経過観察を行い、経過を見ます。
|
| Ⅴ:もやもや病 (Moyamoya disease) |
1:概念
(1)特徴
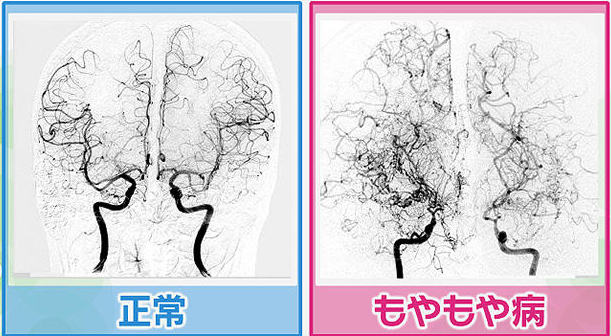
Willis動脈輪閉塞症、とも呼bなれます。
内頚動脈終末部からWillis動脈輪、さらに大脳主幹動脈基部にかけて血管が閉塞をきたす病気です。
X線造影検査で脳低部を中心に細いもやもやとした異常血管網が認められることから命名された疾患です。
大脳前半部の脳血流は相対的に低下しますが、基底核部の脳血流が異常血管網により比較的保たれます。
(2)歴史
西本詮が初めて報告し、1965年に鈴木二郎が命名しました。
(3)原因遺伝子と遺伝形式
3番染色体(3p26-p24.2)にあるMYMY1遺伝子、
17番染色体(17p25)にあるMYMY2遺伝子、
8番染色体(8p23)にあるMYMY3遺伝子が関連していると考えられています。
約10%に家族内発生がみられます。
2:疫学
(1)出生頻度
28万5千人に1人。
3:もやもや病の症候・症状
(1)全身的特徴
基底核部の血管形態などから, 6型に分類されます。
発症年齢により若年型と成人型に分類されます。
症状から、出血型、てんかん型、梗塞型、TIA型などにも分類されます。
小児において、意識障害、脱力発作, 感覚異常,不随意運動, けいれんや頭痛などが生じます。
成人では脳虚血症状や頭蓋内出血による脳卒中発作があります。
(2)合併症
脳梗塞による知的能力低下、失語や全盲などがあります。
4:もやもや病と歯科医療
(1)歯科的特徴
歯科治療中に意識障害やけいれんなどを生じることがあります。
(2)歯科的対応
発作時は, 通常数十分でおきまるまで、静かに寝かせて回復を待ちます。
それ以上回復がみられない場合は、専門医療機関を受診させます。
脳梗塞や脳出血による麻痺があるときは、姿勢に注意が必要となります。
小児では、泣くと一過性虚血性発作が起こりやすいことに注意します。
|
| Ⅵ:レノックス・ガスト(Lennox-Gastaut)症候群 (Lennox-Gastaut syndrom) |
1:概念
(1)特徴
幼児期に発症する年齢依存性てんかん性脳症の一つです。
発症まで正常発達をする潜因性と,さまざまな脳障害を基盤にする症候性に分類されます。
症候性では、West症候群から移行する場合と、発達障害や脳性麻揮など基礎疾患に合併するものがあります。
参照、「てんかん」へ

(2)歴史
1939年にW.G.Lennox (1884~1960)が初めて報告しました。
1966年にH・J.P.Gastaut (1915~1995) らが一つの疾患概念を確立しました。
(3)原因遺伝子と遺伝形式
不明です。
てんかんの家族歴を有するとの報告もあります。
2:疫学
(1)出生頻度
2万人に1人。
3:レノックス・ガスト症候群の症候・症状
(1)全身的特徴
多くは就学前の幼児期に発症します。
多種類の発作をもっており、主要な発作は全般性強直発作が特に重要です。
失立発作や非定型欠神発作があります
発作の回数は多くはてんかん重積状態を伴うこともあります。
連日の発作もめずらしくなく、難治性です。
約60%は症候性です。
(2)合併症
ミオクロニー発作、全般性強直間代発作や部分発作などがあります。
4:レノックス・ガスト症候群と歯科医療
(1)歯科的特徴
抗てんかん薬による歯肉肥大、転倒による歯の破折等が起こりやすくなります。
歯科治療中の発作などがあります。
(2)歯科的対応
てんかんへの対応を行います。
|
| Ⅶ:ウエスト(West)症候群 (West syndrome)
|
1:概念
(1)特徴
点頭てんかんとも呼ばれる、乳児期に発症する年齢依存性てんかん性脳症の一つです。
Lennox-Gastaut症候群と同様、潜因性と症候性に分類されます。
(2)歴史
1841年にWJ.West (1794~1848)が最初に報告しました。
(3)原因遺伝子と遺伝形式
X染色体上のARX遺伝子(Xp21.3) や CDKL5遺伝子(Xp22.3)が原因と報告されています。
2:疫学
(1)出生頻度
1,600人~2万人に1人.。男児がやや多い.
3:ウエスト症候群の症候・症状
(1)全身的特徴
発作(けいれん)の群発が認められます。
3歳児以下にしか現れることの少ない特徴的な持続性の脳波の異常(ヒプスアレスミア)がみられます。
多くに発達の停止や退行を認めます。
難治性てんかん、点頭てんかん.、1~3秒の屈曲性のけいれんがあります。
(2)合併症
知的能力障害があります。
4:ウエスト症候群と歯科医療
(1)歯科的特徴
抗てんかん薬による歯肉肥大、転倒による歯の破折および歯科治療中の発作などがあります。
(2)歯科的対応
てんかんに対応します。
|
| Ⅷ:ゴールデンハー症候群
|
1:概要
(1)特徴
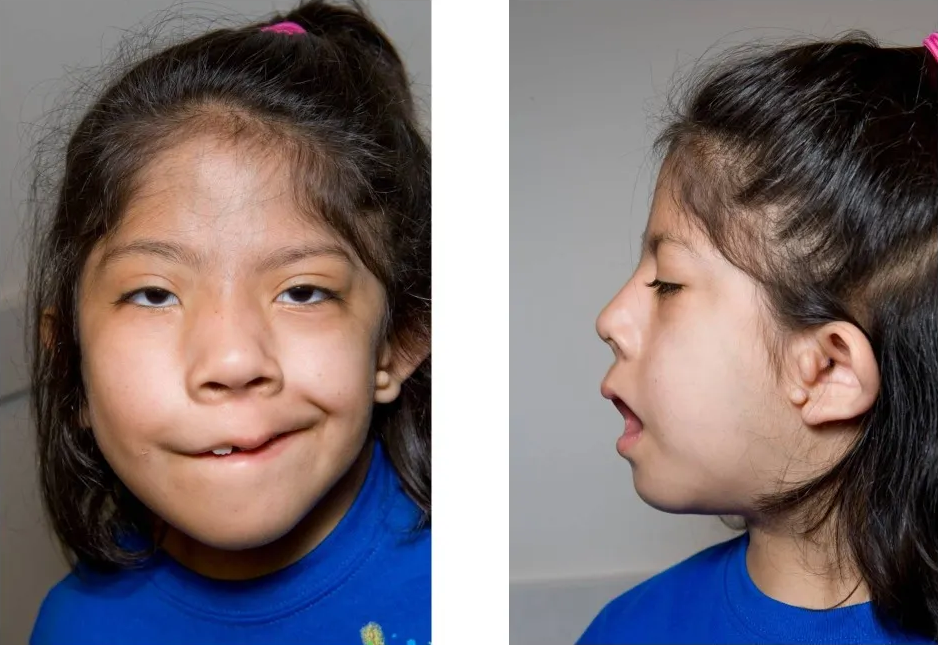
主として第1鰓弓と第2鰓弓の発生異常によって生じます。
耳介・外耳道・顔面の奇形症候群で、左右差のある症状を特徴とします。
小耳症・外耳道閉鎖、眼球結膜類上皮腫、上顎骨・頬骨・下顎骨の低形成、 片側頸椎の完全または部分欠損や
低形成を認めます。
知的障害は伴いません。
2:疫学
(1)出生頻度
ゴールデンハー症候群の有病率は19500~26550人に1人の発生頻度とされています。
軽症例を含めると出生3500~5600人に1人とも報告されています。
最も頻度の高い多発奇形症候群の一つです。
男女比は3:2で、やや男性に多いです。
3:症状
(1)顔面
①頬骨,上顎骨,下顎骨,下顎枝,関節突起,顎関節の形成不全などをきたして顔面非対称を呈します。
②片側の口角部における破裂、すなわち,横顔面裂ないし巨口を示します。
③顔面筋の形成不全と口唇の麻痺。
(2) 耳
①片側性の外耳、中耳の形成不全(小耳症).
②耳珠と口角を結ぶ線上にある皮膚隆起や小耳.
(3)口腔
耳下腺分泌の欠如、舌や軟口蓋の機能障害,交叉咬合.
(4)脊椎
片側性の脊椎,特に頸椎の低形成。
(5)その他
眼球上類皮腫,内耳の障害
口唇裂や口蓋裂,胸椎や腰椎の異常.
|
| Ⅸ:ラッセル・シルバー症候群(RSS)
|
1:概要
(1)特徴
子宮内胎児発育遅延と出生後の成長障害を特徴とする症候群です。
RSSに罹患した小児は、発達遅滞(運動および知的発達)と学習障害のリスクが高いというエビデンスがあります。

(2)原因遺伝子と遺伝形式
第11番染色体上11p15.5領域の低メチル化(エピ変異)によるインプリンティング遺伝子の発現異常、
および第7番染色体母親性ダイソミーを原因として発症します。
2:疫学
本邦で約500~1000名の患者がいると推測されています。
3:症状
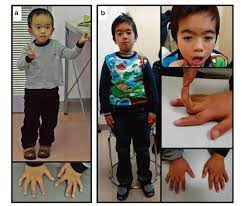
(1)特徴的な症状
典型的には、均整のとれた低身長、正常頭囲、第5指弯曲、額が広く顎が狭い特徴的な三角形の顔、
罹患側の成長障害(片側性発育不全)による非対称な四肢長を有します。
(2)発育障害
罹患乳児の出生体重は一般に平均-2SD以下です。
出生後の成長については、身長が平均-2SD以下です。
RSS罹患児の小児期の成長速度は正常です。
成人の平均身長は、男性151.2 cm、女性 139.9 cmです。
(2)合併症
発達障害、合趾症、胃腸障害、胃食道逆流、食道炎、嚥下障害、新生児期低血糖、心奇形など。
|
| Ⅹ:ポイッツ・イェガース(Peutz-Jeghers)症候群
|
1:概要
(1)特徴
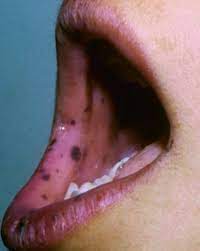
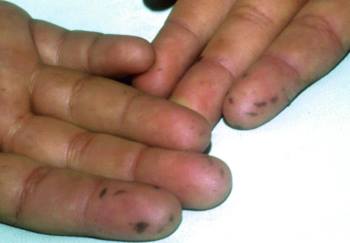
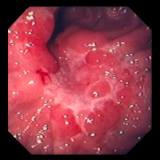
食道を除く全消化管の過誤腫性ポリポーシスと、口唇、口腔、指尖部を中心とする皮膚、粘膜の色素斑を特徴とします。
本症候群でみられる過誤腫性ポリープは粘膜上皮の過誤腫的過形成、粘膜筋板からの平滑筋線維束の樹枝状増生が
特徴であり、ポイツ・イェガースポリープと呼ばれています。
(2)原因遺伝子と遺伝形式
第19番染色体短腕上(19p13.3)に存在するLKB1/STK11遺伝子の突然変異が病因であると考えられていますが、
LKB1/STK11遺伝子変異によりどのような機序で過誤腫性ポリポーシスや色素斑をきたすのかは不明です。
常染色体優性遺伝性疾患です。
発症者の約50%は家族歴がない孤発例です。
2:疫学
国内の患者数は約600~2400人と推計されています。
3:症状
(1)口腔症状
乳児期から口唇、口腔、指尖部などに1~5mmほどの色素斑が認められる。
色素斑は長軸の向きが皮丘、皮溝の流れに一致している。
(2)ポリープ
ポリープは食道を除く全消化管に認め、特に十二指腸から上部空腸に多く認められることが多い。
小児期にポリープ自体の癌化リスクは低い。
しかし、ポリープ増大により、慢性出血による黒色便・血便・貧血、腸重積による急性腹痛・嘔吐、消化管通過障害による
反復性腹痛・嘔吐・便通異常などの症状を呈します。。
幼児期に腸重積を契機に診断される症例もある。
|
| 参考資料 |
|
スペシャルニーズデンティストリー
ダウン症の全てが分かる本
ダウン症ハンドブック

|